
AI for Science
Scientific research serves two major purposes: (i) Discovering fundamental principles, such as the laws of planetary motion and the laws of quantum mechanics, and (ii) solving practical problems, like those that arise in engineering and industry. Researchers typically utilize two major approaches: the Keplerian paradigm (the data-driven approach) and the Newtonian paradigm (the first-principles-driven app-roach). Johannes Kepler’s discovery of the laws of planetary motion is the best example of the former. Kepler found these laws by analyzing experimental data; later, using the laws of mechanics and gravitation, Isaac Newton was able to reduce the problem of planetary motion to ordinary differential equations (ODEs) and derive Kepler’s laws. In short, Kepler first made the discovery but did not understand the reasons behind it, so Newton took it one step further and discovered the fundamental principles — which are applicable to many other problems.
For practical purposes, the task of finding first principles was basically accomplished with the establishment of quantum mechanics. In 1929, Paul Dirac declared that “The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble” [1]. His comment applies not just to chemistry but also to biology, materials science, and numerous other scientific and engineering disciplines. In practical situations, we can often use simplified principles—such as Euler’s equations for gas dynamics and the Navier-Stokes equations for fluid dynamics—instead of relying on quantum mechanics.
With these fundamental principles in hand, essentially all natural science and related engineering problems reduce to mathematical problems — specifically to ODEs or partial differential equations (PDEs). Unfortunately, before the development of effective tools, the only thing that scientists can do to solve these practical problems is to simplify or ignore the principles.
The first major advance in this realm occurred when John von Neumann recognized that computers and numerical algorithms should allow us to directly utilize these fundamental principles in a practical way. Researchers have since developed many numerical algorithms—such as the finite difference method, finite element method, and spectral methods—to solve the corresponding PDEs. The basic starting point of these algorithms is the fact that general functions can be approximated by polynomials or piecewise polynomials. The corresponding impact has been tremendous. Scientific computing has become the foundation of modern technology and engineering science; in fact, the introduction of numerical algorithms has revolutionized countless disciplines, from structural mechanics and fluid mechanics to electromagnetism.
However, many problems cannot yet be treated in this way. For instance, we are still quite far from employing first principles to successfully address material properties, materials design, and drug design. In these types of areas, theoretical work is usually fairly detached from the real world; real-world problems must instead be solved empirically (by trial and error).
All of these “hard” problems share one common factor: they depend on many variables and thus suffer from the curse of dimensionality. For example, consider Schrodinger’s equation in quantum mechanics. Neglecting symmetry, the number of independent variables for the wave function is three times the number of particles. A system with 10 electrons is extremely simple, but a PDE in 30 dimensions is highly nontrivial.
It is here that deep learning might be able to help. Deep learning successfully classifies images, generates fake pictures of human faces, and produces Go strategies that defeat the best human players. While these scenarios are common applications of artificial intelligence (AI), they respectively focus on approximating functions, approximating and sampling probability distributions, and solving Bellman’s equations — all of which are standard problems in applied mathematics. However, these AI problems have much higher dimensions than standard applied math problems.
Deep learning’s effective performance on these problems suggests that deep neural networks are particularly successful at approximating functions in high dimensions. While a complete mathematical theory for deep learning does not yet exist, we do have some hints as to why this is the case. If we approximate a function with piecewise linear functions on a regular mesh, the error is proportional to the square of the mesh size; this is the origin of the curse of dimensionality. But if we instead approximate the function with neural network functions, we can show—at least in some situations—that the error rate does not deteriorate with dimension [4].
This observation has several important implications. Because functions are among the most basic mathematical objects, a new tool that approximates them in high dimension will impact many different areas. In particular, deep learning can help solve the aforementioned problems that suffer from the curse of dimensionality. This is the starting point of AI for science.
The most successful example in this direction is AlphaFold: an AI program from Google DeepMind. By exploiting protein sequence datasets and the most advanced deep learning models, DeepMind shocked the world by developing the AlphaFold 2 algorithm that elegantly solved the protein structure problem [6].
Although AlphaFold 2 is solely driven by data, AI for science is not a purely data-driven paradigm. In fact, the main difference between AI and science is the presence of a set of first principles in science (as discussed earlier). One major component in AI for science is the use of AI to develop better algorithms or approximate models for these first principles. In this regard, the best-known example occurs in molecular dynamics — a fundamental tool in biology, materials science, and chemistry. The idea is to study the properties of molecules and materials by examining the dynamic trajectories of the atoms in the system (the dynamics of the atoms simply follow Newton’s law). The difficult part involves modeling the forces between the atoms, which are governed by the interatomic potential. In the past, scientists have either tried to guess an approximate functional form of the interatomic potential (the empirical approach) or used quantum mechanics models to compute the forces on the fly (the ab initio approach). The former is unreliable and the latter is quite expensive.
Machine learning offers a new paradigm that only uses quantum mechanics to supply the data. Based on that data, we can employ machine learning to create an accurate approximation of the interatomic potential, then utilize that approximation to perform molecular dynamics simulations.
To truly make this technique work, we must address two important issues. The first issue pertains to network structure, which should be extensive and respect physics. Being extensive allows us to perform learning on small systems and use the results for larger systems; respecting physics means that we need to keep the symmetries, conservation laws, and other physical constraints in place. In the current context, the main problem involves maintaining the translational, rotational, and permutational symmetries [8].
The second issue concerns data. If we ultimately seek an approximate potential energy function that performs as well as the original quantum mechanics model in all practical situations, the training dataset must be able to represent these situations. However, we want the dataset to be as small as possible since the quantum mechanics computations that calculate the data are expensive. This conundrum calls for an adaptive data generation scheme—such as the exploration-labeling-training algorithm—that generates data on the fly as learning takes place [9].
Given these requirements, we can indeed produce neural network approximations of the interatomic potential with ab initio accuracy for a large class of (if not all) atomic systems. One relevant model is called Deep Potential Molecular Dynamics (DeePMD) (see Figure 1). Together with high-performance computing, DeePMD has extended our ability to perform molecular dynamics with ab initio accuracy on systems with thousands of atoms to systems with billions of atoms [3, 5]. The DeePMD software package DeePMD-kit facilitates the use of DeePMD with minimal effort [7].
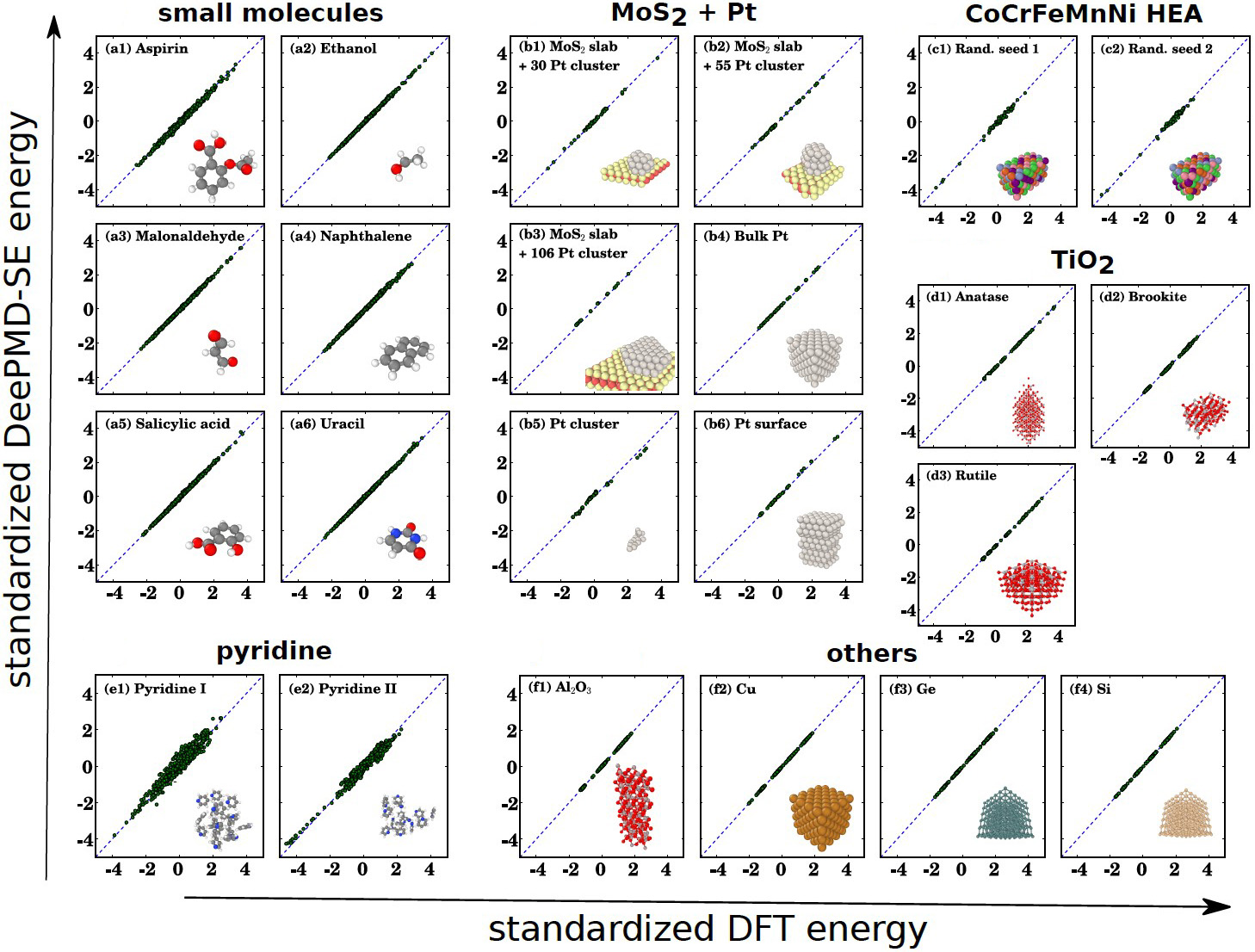
We can apply similar ideas to other modeling schemes. For example, we can utilize highly accurate quantum chemistry computational data to train density functional theory models that are more universal and accurate. We can also develop more accurate and reliable coarse-grained molecular dynamics models, more accurate moment closure models for kinetic equations, and so forth. In fact, machine learning serves as the missing tool for multiscale, multiphysics modeling.
AI techniques can also enhance our experimental capabilities by providing inversion algorithms that are more efficient and accurate. AI-based algorithms deliver realistic and accurate data for the forward problem, and we can exploit the differentiable structure in AI-based formulations to solve the inverse problem. This line of work is still at a preliminary stage, but it will undoubtedly change the way in which experiments are conducted and experimental apparatuses are designed.
In addition, AI will likely have a serious impact on the handling of literature and other scientific knowledge. These sources are major suppliers of inspiration for our research, but finding and studying them is a highly nontrivial process. We can thus imagine the use of AI—such as AI databases and large language models—to collect and query the existing literature in a more efficient manner.
With these possibilities comes a new “Android paradigm” for scientific research. In this novel paradigm, the scientific community will work together to build an infrastructure that includes AI-based algorithms for physical models, AI-enhanced experimental facilities, and an innovative knowledge database that encompasses the literature and other scientific data. These platforms constitute the “Android platform” for scientific research. Scientists can then organize work on specific applications—like the search for catalysts in a particular reaction or the design of new batteries—on top of this Android platform. This horizontally integrated approach should undoubtedly accelerate the process of scientific research, help break disciplinary barriers, and enhance interdisciplinary research and education.
With this vision in mind, we initiated the DeepModeling open source platform in 2018. This platform invites the scientific community to work together and build the AI-enhanced infrastructure for physical modeling and data analysis. It has since attracted hundreds of developers and facilitated more than 40 projects under active collaboration.
Such a paradigm shift will also bring fundamental changes to the field of applied mathematics. After all, applied math has always centered on the development of tools for first-principles-driven and data-driven approaches [2]. The necessary ingredients for the Android platform are also the major components of applied mathematics. As science becomes increasingly integrated under this platform, applied math will likely become the foundation of interdisciplinary research.
References
[1] Dirac, P.A.M. (1929). Quantum mechanics of many-electron systems. Proc. R. Soc. Lond. A, 123(792), 714-733.
[2] E, W. (2021). The dawning of a new era in applied mathematics. Not. Am. Math. Soc., 68(4), 565-571.
[3] E, W., Han, J., & Zhang, L. (2021). Machine learning-assisted modeling. Phys. Today, 74(7), 36-41.
[4] E, W., Ma, C., Wu, L., & Wojtowytsch, S. (2020). Towards a mathematical understanding of neural network-based machine learning: What we know and what we don’t. CSIAM Trans. Appl. Math., 1, 561-615.
[5] Jia, W., Wang, H., Chen, M., Lu, D., Lin, L., E, W., … Zhang, L. (2020). Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. In SC ’20: Proceedings of the international conference for high performance computing, networking, storage and analysis. IEEE Press.
[6] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583-589.
[7] Zeng, J., Zhang, D., Lu, D., Mo, P., Li, Z., Chen, Y., … Wang, H. (2023). DeePMD-kit v2: A software package for deep potential models. J. Chem. Phys., 159(5), 054801.
[8] Zhang, L. Han, J., Wang, H., Saidi, W., Car, R., & E, W. (2018). End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems. In Advances in neural information processing systems 31 (NeurIPS 2018). Montreal, Canada. Curran Associates, Inc.
[9] Zhang, L., Wang, H., & E, W. (2018). Reinforced dynamics for enhanced sampling in large atomic and molecular systems. J. Chem. Phys., 148(12), 124113.
About the Author
Weinan E
Director, AI for Science Institute in Beijing
Weinan E is director of the AI for Science Institute in Beijing and a professor in the Center for Machine Learning Research and the School of Mathematical Sciences at Peking University. His main research interests are in numerical algorithms, machine learning, and multiscale modeling with applications to chemistry, materials science, biology, and fluid mechanics.
